年前CJ大佬深度体验了下WGCNAshiny, 提了一些非常好的建议和意见,过年的时候折腾了几天,改善了一些东西,距离上次发布更新已经过去很久了,期间随着课题的进展,我自己也添加了一些内容。
太长不看省流版本
安装方法:
- 通过TBtools插件:新插件更新-TBtools 直接安装打包好的插件即可,非常方便!
- Rstudio ShawnWx2019/WGCNA-shinyApp , 更新shinyapp之前,通过下面代码更新依赖包.
devtools::install_github("ShawnWx2019/WGCNAShinyFun",ref = "master")
更新内容:
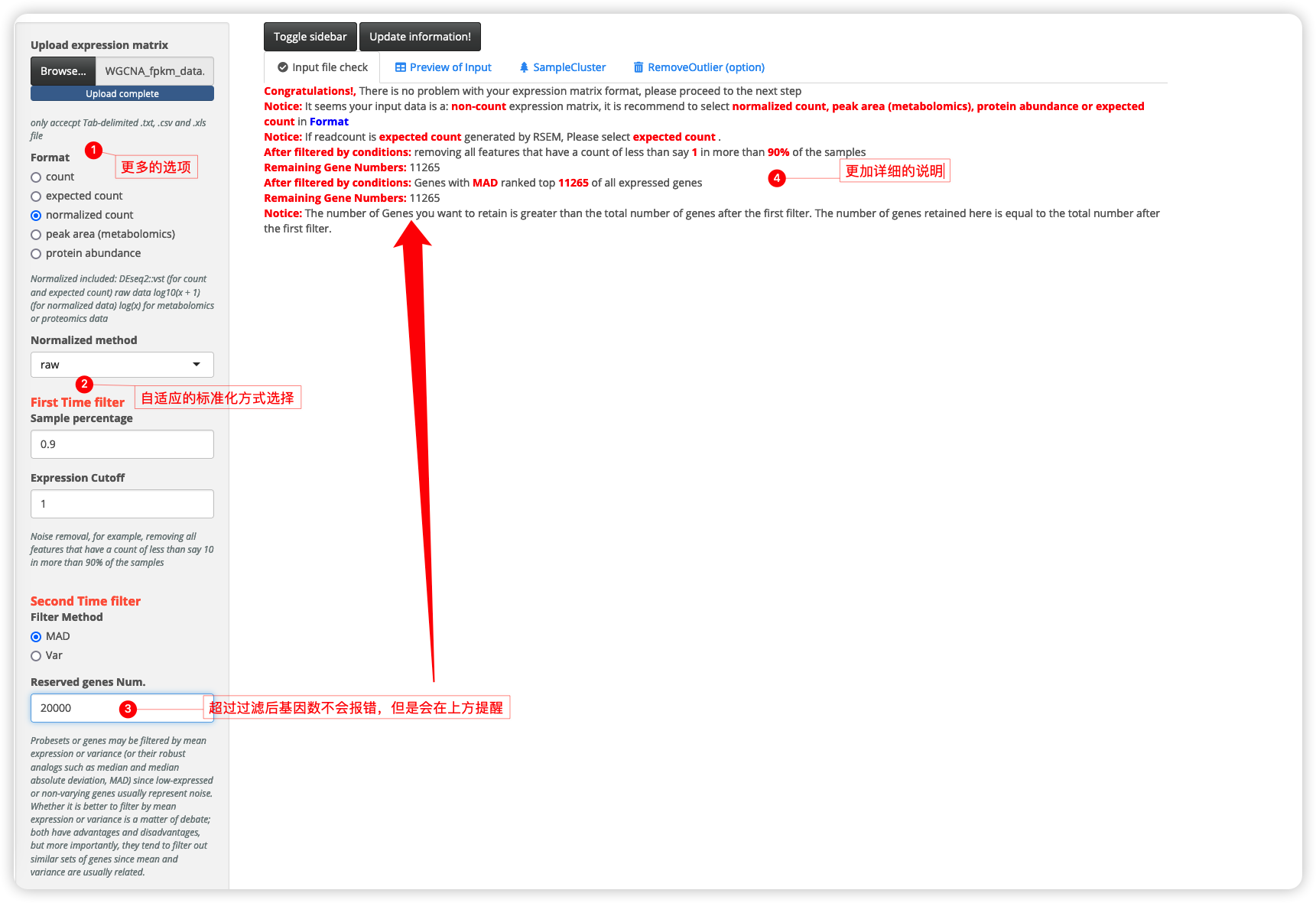
- 🔨 改动了表达量矩阵输入类型。从之前的
count和FPKM更新为count、expected count、normalized count、peak area以及protein abundance。更加准确和符合逻辑; - 🍭 保留基因数量(reserved genes number)超过过滤后的基因数量会自动选择过滤后的基因数量;
- 🎊 去除离群样本(Remove outlier);
- 🎊 在网络构建和模块-性状关系构建步骤增加progress bar,防止多次点击导致程序重复运行;
- 💪加入迭代WGCNA(iterative WGCNA)选项,开发中.... 不过可以通过
method2的方式净化模块。 - 🍭 参数输出,这里增加了
Env output选项卡,方便有R语言基础的同学保留shinyapp运行时使用的参数配置,结合R语言进行进一步分析,同时方便复现之前的结果。
啰嗦版本
为何要加入代谢组和蛋白组数据
细心的同学注意到我在新版本中输入数据的format有了很大改动。当然之前的count和FPKM太过随意了。下面从两部分解释下为何要修改成目前这样,当然也广泛接受大家的意见和建议,会在下个版本继续更新。
首先整体其实还是分了两大块,总结一下就是read count类型数据和其他类型数据。
| 格式 | 特征 | 备注 | 标准化方法 |
|---|---|---|---|
| count | 整数 | vst | |
| expected count | 不全是整数 | 通过向上取整的方式转换为整数 | vst |
| normalized count | 归一化后的readcount | Fpkm tpm cpm rpkm 等 | Raw/log10(x+1) |
| peak area(metabolomics) | 一般数值会非常大 | 不推荐直接使用原始峰面积,需要使用标准化后的数值,比如商业软件CD,开源软件tidymass等进行数据过滤和归一化后的代谢物积累矩阵 | Log10(x) |
| protein abundance | 同样需要归一化以后的蛋白质丰度。 | Log10(x) |
这里值得注意的是我把非靶代谢和蛋白质组学的数据也放进来了,这并不意味着这类型的数据无论从数学原理出发还是生物学原理出发适合构建共表达网络。
我这里将非靶代谢组学的数据和蛋白质组学数据加入进来,本意是并不是非要通过WGCNA构建共表达网络,而是将WGCNA作为一种高效数据挖掘的方法。 例如当我们在做大规模(large-scale)非靶代谢组学数据分析时,会发现我们会面临很多问题,比如对自然群体的非靶代谢组学分析,如果通过k-means,hclust等方法去归纳群体材料和代谢物类型之间的关系会很吃力,但是同过WGCNA将积累规律类似的代谢物进行模块划分,有很大几率总结出材料-代谢物之间的规律,提高了数据挖掘的效率。而后续的代谢物共积累网络构建则成为锦上添花的内容,并非重点。后续有机会可能会展开讲一下想法,不一定准确。
UI改动:
代码改动:
getdatExpr = function(rawdata,RcCutoff,samplePerc,datatype,method){
##> Noise filtering
x <-
rawdata %>%
setNames(c("id",colnames(.)[-1])) %>%
mutate(id = as.character(id)) %>% ## aviod numeric columns.
column_to_rownames("id") %>%
filter(
rowSums(. > RcCutoff) > (samplePerc*ncol(.))
)
if (
datatype == "expected count"
) {
x <- x %>%
mutate_if(is.numeric,ceiling)
}
##> Normalization
if(method == "vst") {
dx <- x %>%
as.matrix() %>%
varianceStabilizingTransformation(., blind = TRUE)
} else if (method == "logarithm") {
if (datatype == "normalized count") {
dx <- log10(x+1)
} else {
dx <- log10(x)
}
} else if (method == "raw") {
dx <- x
} else {
return()
}
return(dx)
}
程序防呆设计
这些错误可以忽略掉了,放心造
- 规避纯数字基因名称 根据用户的反馈,发现某些转录组数据的gene id是纯数字的,在R对纯数字的列转换成rownames如果不连续会报错,所以这里会把第一列直接转换成字符格式,防止该错误发生。
- 自动转换标准化方式,上面介绍了不同数据类型有对应的标准化方法,当然我知道不是每个用户都会认真读手册,所以这里做了自适应,在
format确定了格式后,Normalized method只会显示对应的标准化方式选项。 - 保留基因数量大于了过滤后总数 这个也是常见的报错,如果你填写的
reserved gene Num.大于了过滤后的基因数量会导致报错,这里加了判断,如果这种情况发生,直接保留过滤后的基因数目。
进度条
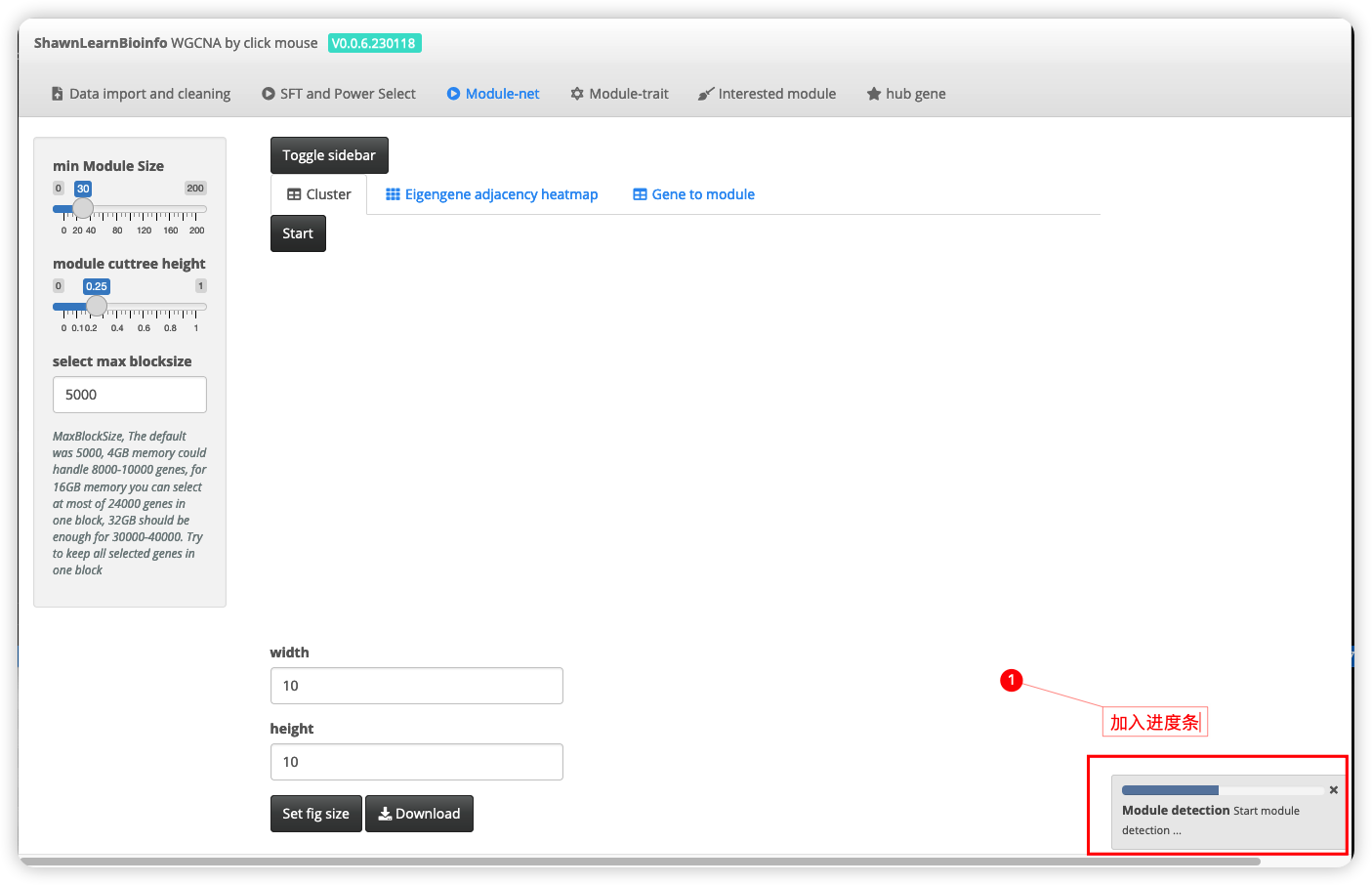
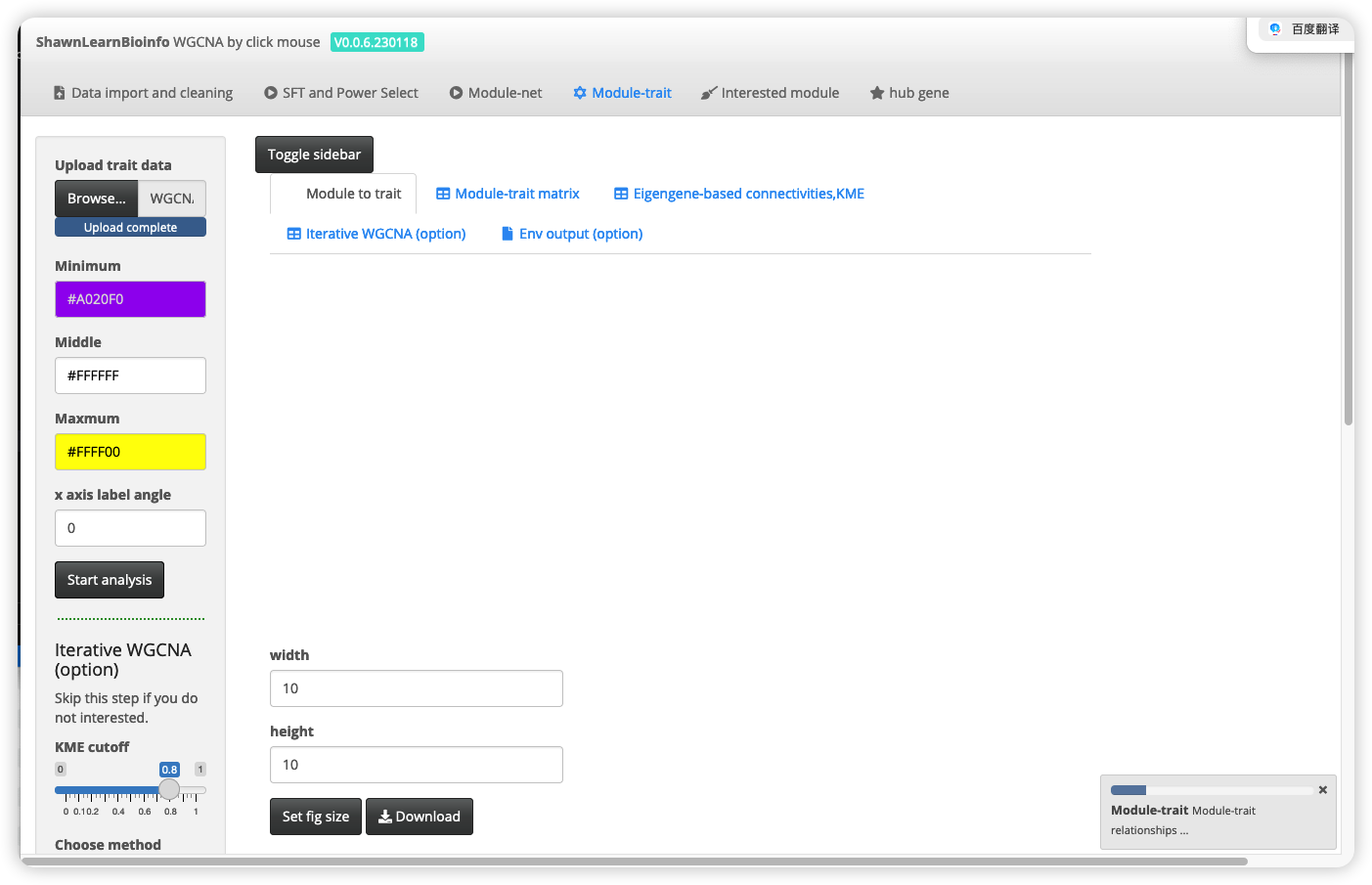
CJ大佬在体验的时候发现在等待时间较长的Module-net阶段,不知道运行完没有,有些用户可能会觉得是不是没点到start 于是就又按了几次,特别是对于没有更新新版本Rserver以及Rstudio用户中没有折腾blas的用户,这个等待时间会更长,所以现在在module-net和module-trait加入了和SFT步骤中一样的进度条,你点了就会跳progress bar运行完消失。
UI画面:
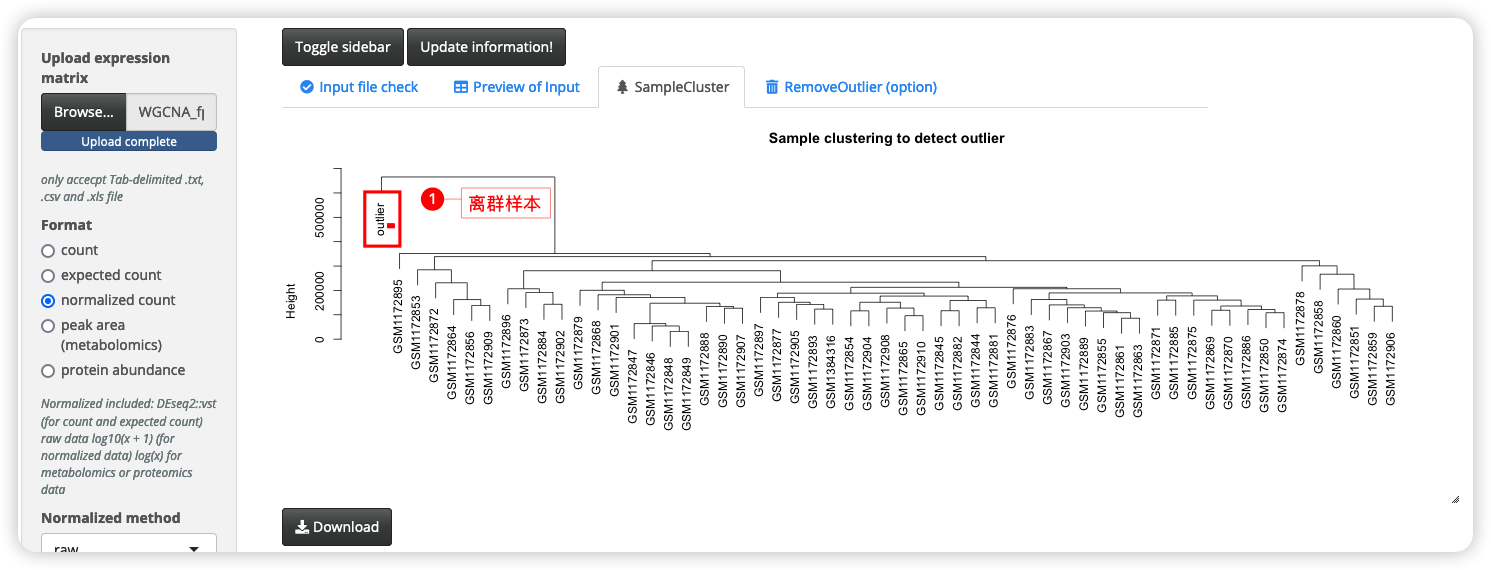
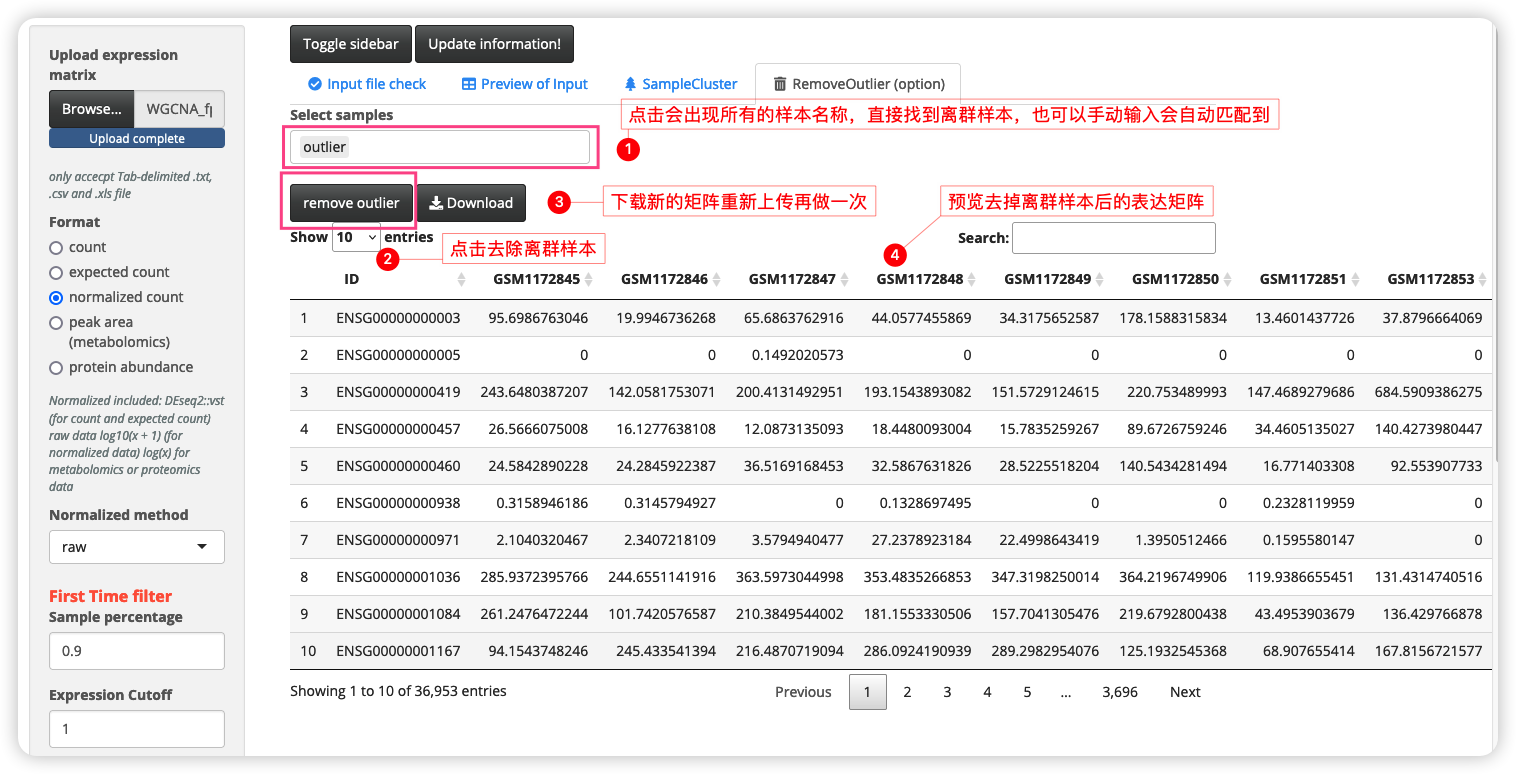
去除离群样本
有时候数据量大的时候可能会出现离群样本,如下图,我们可以通过RemoveOutlier(option)来去掉离群样本,由于shiny中比较难实现do-over这样的过程,所以就得麻烦大家重新开始了,**开始上传前最好刷新下网页,并把鼠标点到第一个选项卡input file check。然后再点update information
迭代WGCNA
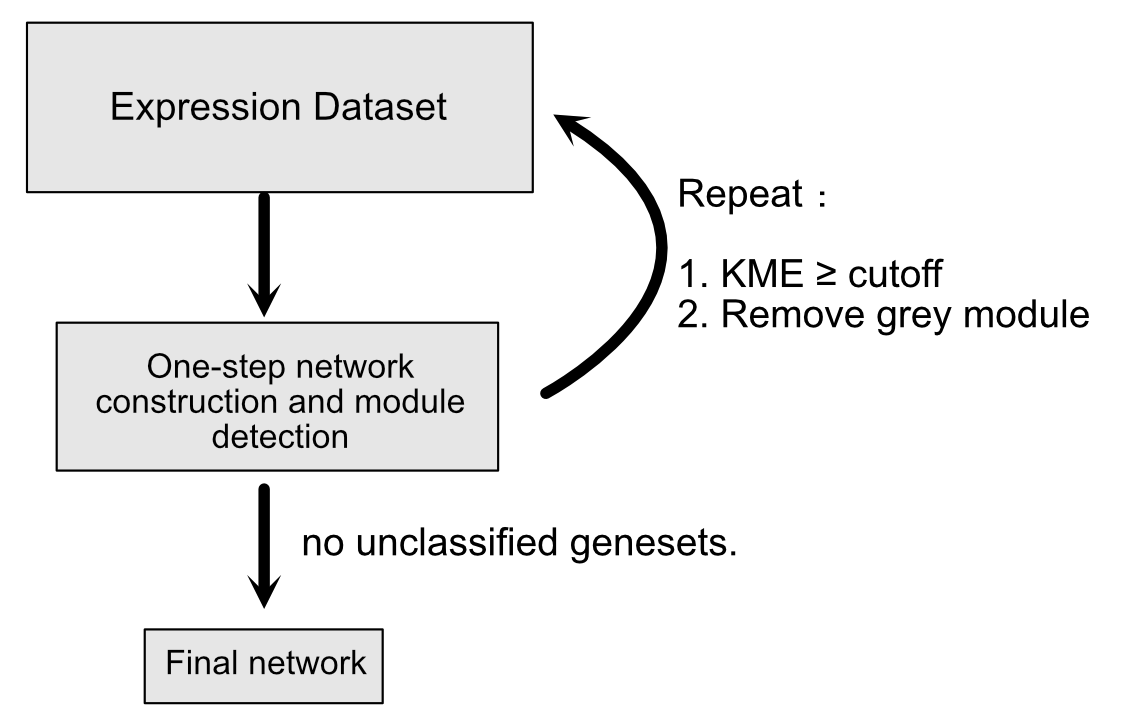
关于迭代WGCNA,推荐看iterativeWGCNA: iterative refinement to improve module detection from WGCNA co-expression networks 作者用python搭建的流程,有需要的可以通过cstoechert/iterativeWGCNA 实现。这部分我并没有用shinyapp复现 确实难度比较大,加上之前说的do over很难,基本做不到自动迭代。所以method1是没有用的。可能后续有精力了尝试折腾下。
这里method2提到的迭代,原理和方法和上面是不一样的,我的目的也很明确,就是首先在可以获得到一个非常趋近于无标度网络的情况下(有非常合适的软阈值),这个网络首先是比较稳健的。然后在此基础上去掉无法分类的基因和一部分kME(eigengene based connectivity)较低的基因来纯化模块,迭代过程中sft可能发生细微的变化,但整体不会发生很大的变化。这样无论在数据挖掘还是后续的共表达网络可视化都会有一定程度的优化。
基本操作就是
基本原理:
方法:
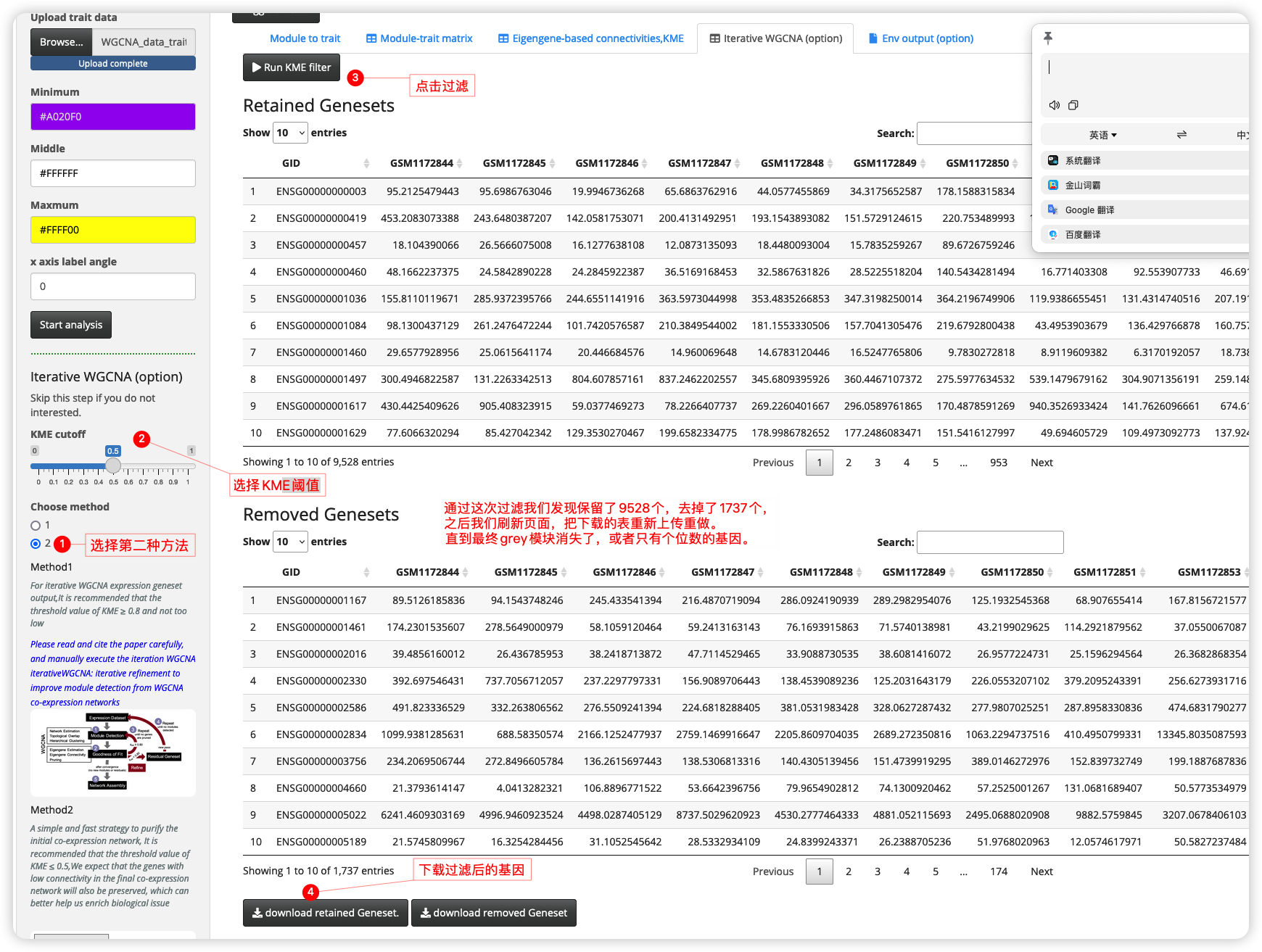
项目示例:

环境变量导出
经常遇到这种问题,过了很长时间,想回来复现一下当时通过WGCNAshiny跑的结果,发现怎么也对应不上,后来发现是当时设置的参数忘记了,这里加入了Env output,当做完module-trait之后会看见该选项卡,点击数据整合Integrate data会立刻把所有中间变量以.rsd的形式保存下来,然后点击下载可以得到一个EnvImg.rsd的文件。这个可以在Rstudio中通过readRDS('filepath/EnvImg.rsd') 加载出来,这样不仅可以看到当时所有的参数,而且运行数据所有的中间变量都被保存,有R基础的可以直接拿去进行下游分析。
比如我们把EnvImg.rsd放在家目录下的Download文件夹下
x <- readRDS("~/Downloads/EnvImg.rds")
# 我们通过x$ 就可以查看参数和中间变量
x是一个列表list,存放这所有的中间变量和参数

最后
多谢CJ大佬提意见,还有群里的老铁们不断鞭策,鸽王又鸽了大家一回!😂
