MetMiner流程整合了mass_dataset数据输入格式,所以具有灵活多变的数据输入模式,本文介绍了通过metMiner进行代谢组数据分析前的数据准备及项目初始化工作。
主要从一下几方面说明:
- Metadata准备(实验数据管理及样本命名规则);
- 原始数据格式转换和文件结构(从原始数据导入);
- 提峰表格准备(从其它软件提峰表格导入);
- mass_dataset准备(从mass_dataset类型数据导入);
1. 实验数据准备和样本命名规则
为了对接MetMiner分析流程,在实验时候需要严格遵守样本命名规则,这样后续的分析流程才会顺畅,不容易报错。
原因是在数据处理时,样本名称有时候会被当做row names 或者 column names,在用bash脚本或者R语言处理文件时,一些特殊符号会造成报错,而我们又不可能耗费巨大的经理去写海量的判断语句来甄别这些错误,所以我们在数据分析流程端采用特定的命名规则,通过meta data来规范样本名称。
规范化的元数据记录可以帮助我们更好的管理数据。
1.1 准备元数据(meta data)
元数据表格一般包含以下内容
- 原始样本名称(sample name raw): 这个是实验前期对于采样样本的原始命名,例如
col0-leaf-1 max2-leaf-1,这个按照自己的理解以及一定的命名规则对各个样本进行标记,这里不做要求,但尽量避免用中文,因为可能有些机器在处理中文字符时可能出现乱码; - 样本类型(class): QC样本标记为大写的**
QC,测试样本标记为Subject**,S大写; - 规范样本id(sample_id):这个id是我们在数据处理时对样本的编码,需要遵循以下规则:
- 对于QC(quality control)样本,以大写的**
"QC_"**开头, - 对于测试(Subject)样本,以大写的**
"S_"**开头, - 数字编号补齐:
QC_或者S_后面接序号,根据QC样本的数量增加pad 0,意思是如果QC的数量为两位数,那么在QC 1-9的命名需要再1-9前面添加一个0即:QC_01,QC_02,QC_03..., 同样如果QC样本的数量为3位数,需要在1-9前面加两个0,在10-99前面加一个0,即QC_001,QC_002,QC_003, ...QC_010。原因是QC_xx在excel以及R时字符型数据,在排序时,会根据第一个检测到的数字进行排序,如果不进行补零填充,那么对QC序列进行排序时顺序会变为QC_1,QC_10,QC_100QC_1000,QC_1001,在后续可视化或者分析过程中可能会出错;同样对于Subject样本编号方式也是如此。
- 对于QC(quality control)样本,以大写的**
- 样本分组(group):样本所属的组别,对于设置了生物学重复的样本,需要表明每个样本所属的组别,比如
case,control,treat,mock; - 进样顺序(injection.order): 根据样本实际上样顺序标记,这里为纯数字形式;
- 其它信息:如果对样本包含的变量非常丰富,可以依次增加信息,比如
tissue,treat,day,gender,species这些信息的加入,在数据清洗及数据挖掘步骤,可以通过选择这些属性来专注这些单一变量带来的代谢水平差异。对于QC样本这里可以不标注,空着即可,也可以标注为QC。
2. 数据准备
首先介绍从LC-MS下机数据开始处理。
2.1 选项1 - 原始数据格式转换
获得LC-MS下机数据后,需要将原始数据转换为.mzxml以及.mgf格式,以Thermo QE Plus下机数据为例,获得.raw文件后通过proteowizard提供的msConvert进行转换。整体数据格式转换过程可以参考TidyMass提供的教程:
对于DIA采集的数据可能需要其它的软件导出.mgf文件,例如waters MSe需要通过UNIFI软件导出
2.2 原始数据文件路径结构
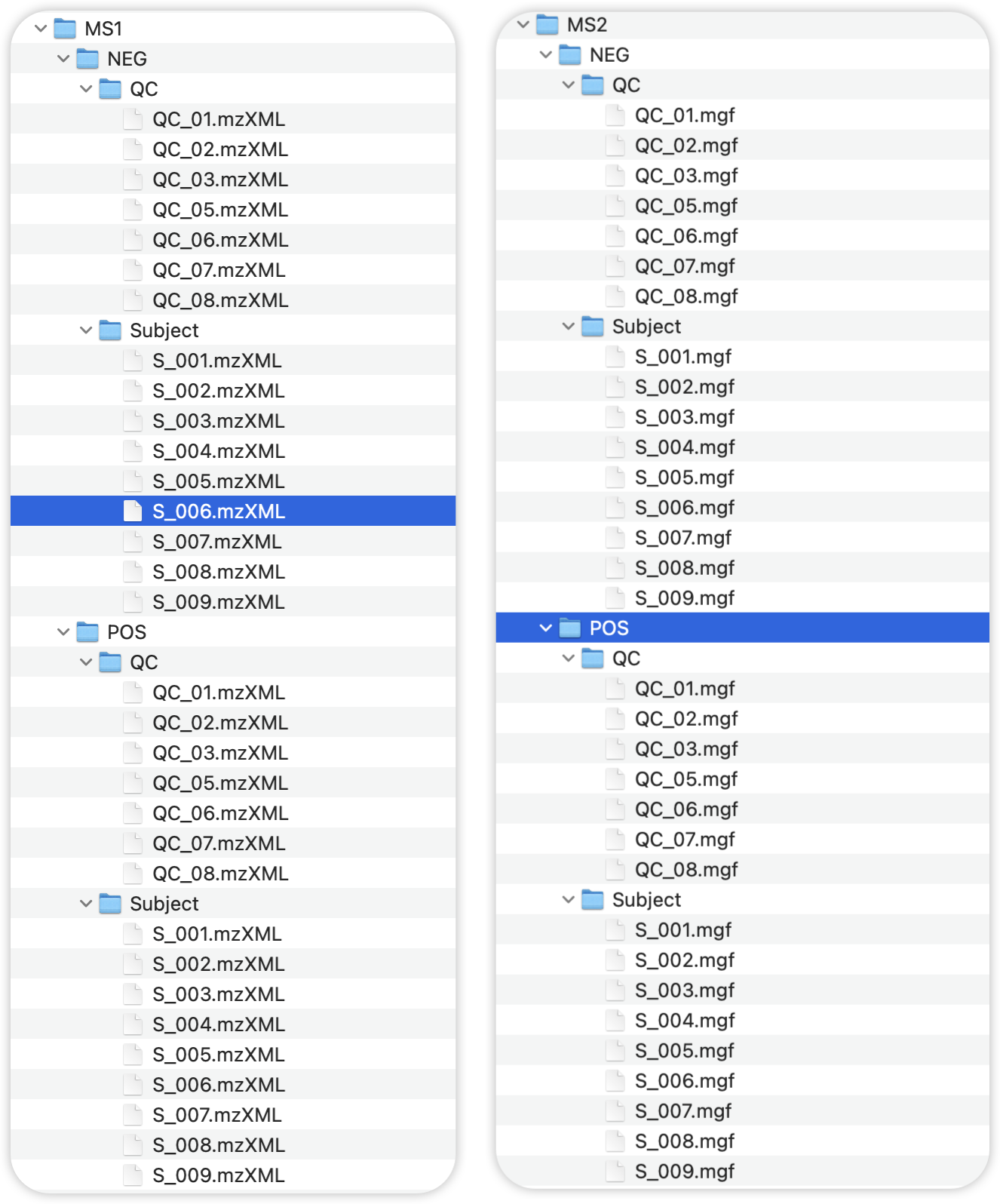
我们在 1.0 的部分建议的命名规则对数据进行整理,然后按照下图整理文件,文件夹的名字也必须和图片中一致。
MS1 文件结构 MS1/MS2文件夹下分NEG和POS两个子文件夹,而后分别将QC和Subject的.mzxml或者.mgf格式的文件放到对应的文件夹下。
MS1
├── NEG
│ ├── QC
│ └── Subject
└── POS
├── QC
└── Subject
MS2
├── NEG
│ ├── QC
│ └── Subject
└── POS
├── QC
└── Subject
2.3 选项2 原始提峰表格
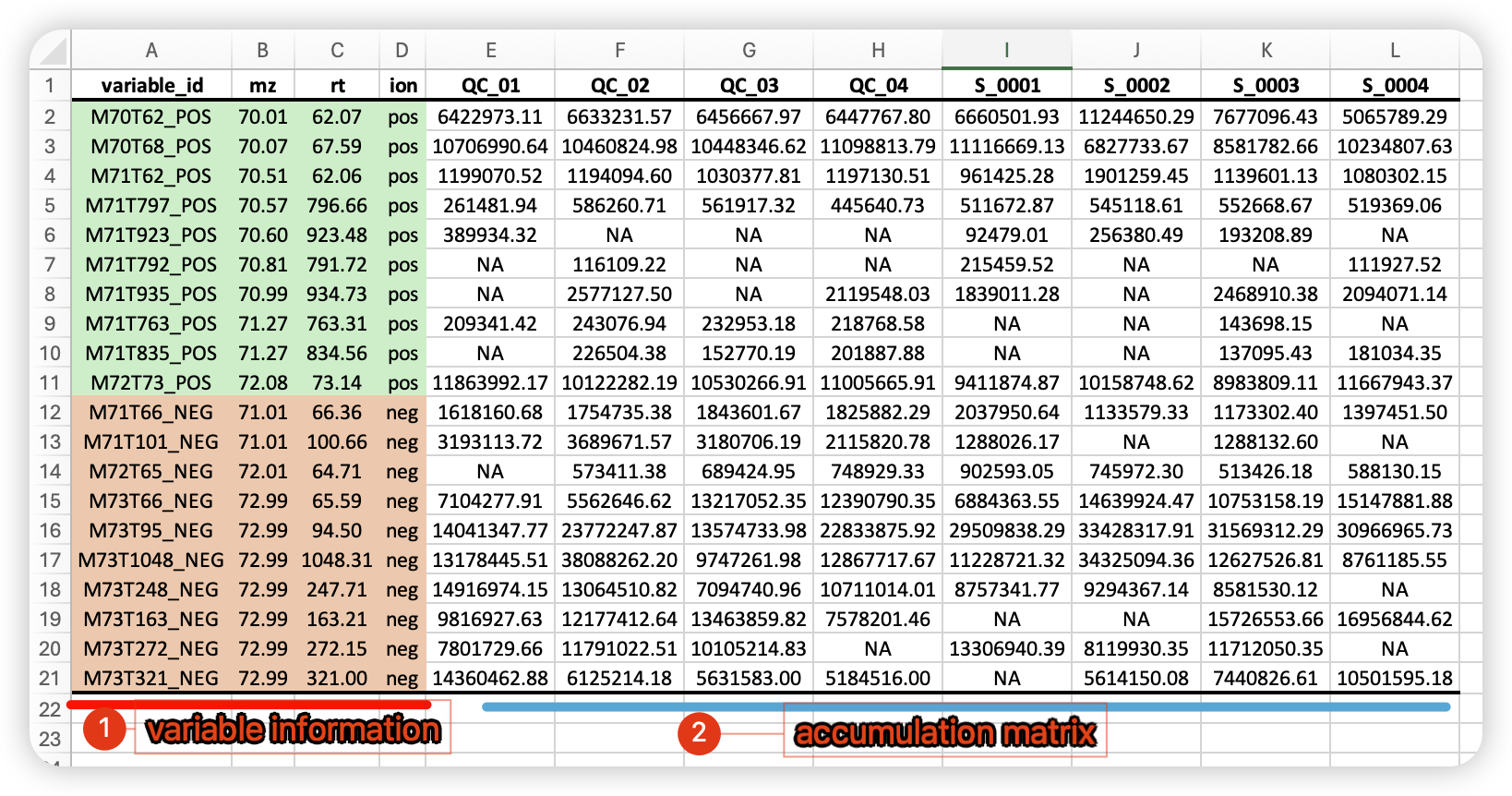
当我们从其它提峰软件获得提峰表格后,将feature的MS1信息以及提峰表格信息整理成下表,
其中前4列为variable information 尽量保证列名和图中一致,代谢物id,质荷比,保留时间,离子模式。后面为QC和样本中峰面积。
2.4 选项3 mass_dataset object
我们用TidyMass软件处理过数据后如果想重新分析,通过save()将mass_dataset数据保存为.rda后缀的文件。
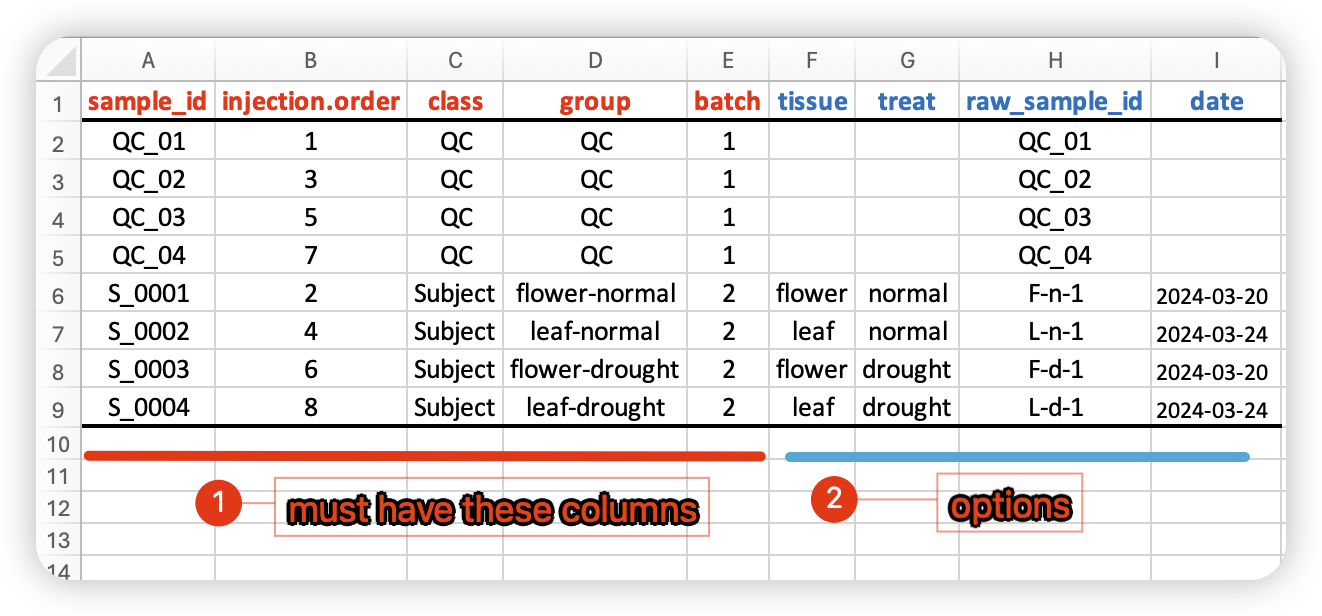
2.5 样品信息表 (必须)
基本和metadata中的信息一致 ,但是要注意添加了injection.order和batch两个信息, 这两个信息在数据清洗时用来校正系统误差。如果不确定是否存在批次效应,batch可以全部填1,后续如果检查出批次效应,在回来校正该数据。
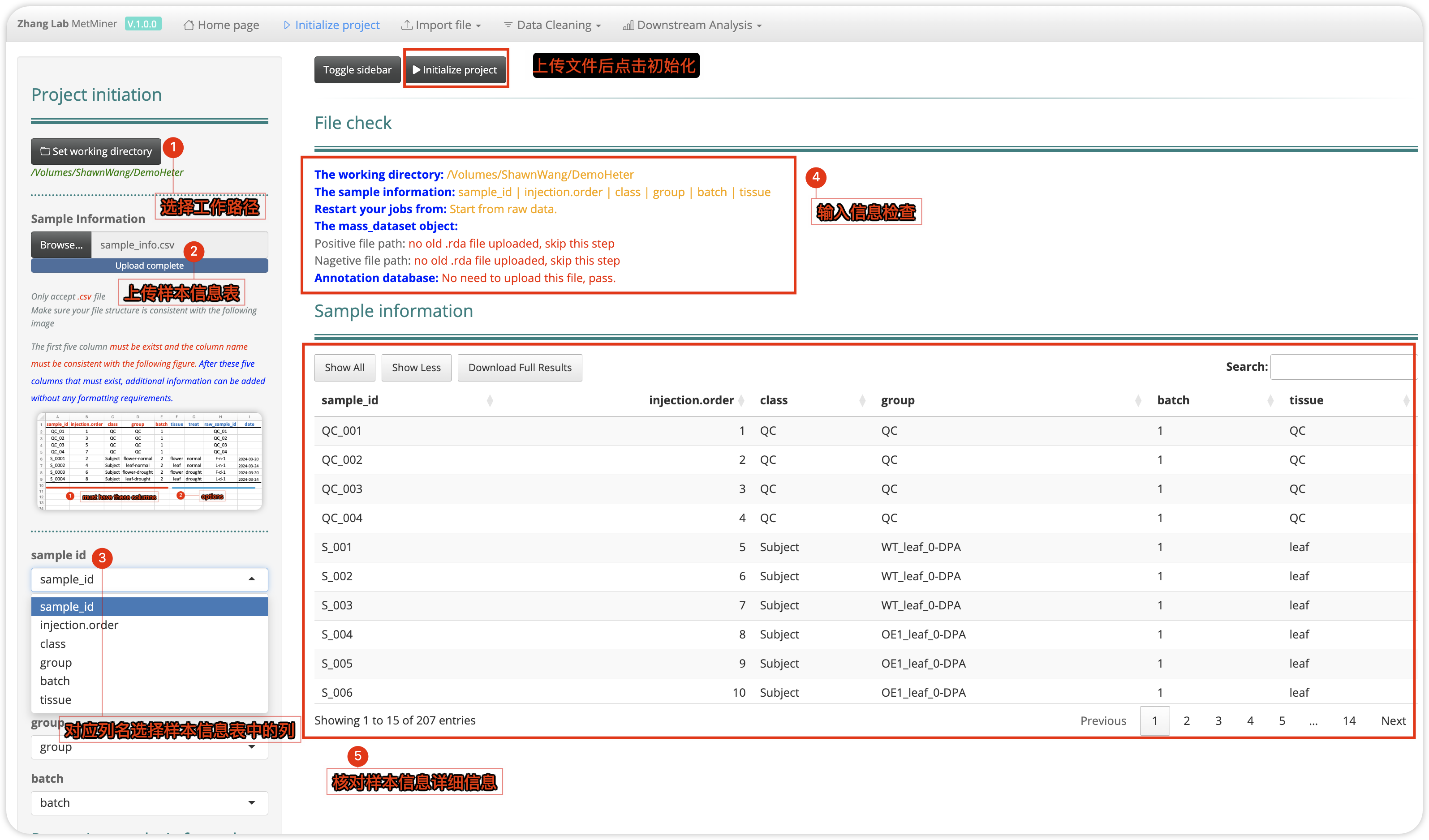
3. 项目初始化
首先我们需要选择一个路径(文件夹)为项目的工作目录,最好将输入数据都放到该文件夹下,同时运行过程中需要保存的数据会自动保存到该路径下。
其次需要将样本信息表格上传,上传后在左侧sidebar处会自动提取上传表格中的列名,如果顺序和软件默认顺序以及列名和默认列名不匹配,需要对前5列信息一一对应匹配,然后软件会校正统计表格;
确认好后点击initialize project项目会初始化,file check会返回工作目录的路径,sample information中包含的信息。
File check
----------------------
The working directory: /Volumes/ShawnWang/DemoHeter
The sample information: sample_id | injection.order | class | group | batch | tissue
Restart your jobs from: Start from raw data.
The mass_dataset object:
Positive file path: no old .rda file uploaded, skip this step
Nagetive file path: no old .rda file uploaded, skip this step
Annotation database: No need to upload this file, pass.
Sample information部分会将初始化后的样品信息表格打印出来,详细检查下对应关系,确保没有问题,可以进行下一步。
---The end---
Jun 22, 2024 by Shawn Wang, HENU, Kaifeng, Henan, China.
