MetMiner流程整合了mass_dataset数据输入格式,所以具有灵活多变的数据输入模式,本文介绍了通过metMiner进行代谢组数据分析前的数据准备及项目初始化工作。
主要从一下几方面说明:
- 原始数据格式转换和文件结构(从原始数据导入);
- 提峰表格准备(从其它软件提峰表格导入);
- mass_dataset准备(从mass_dataset类型数据导入);
原始数据格式转换
这里以ThermoFisher的Q Exactive™ Plus产生的数据为例, LC-MS产生的原始数据为.raw格式,需要通过ProteoWizard-MSCovert软件转换为包含MS1信息的mzXML格式和包含MS2信息的mgf格式。windows用户可以直接在界面软件按照下图进行格式转换:
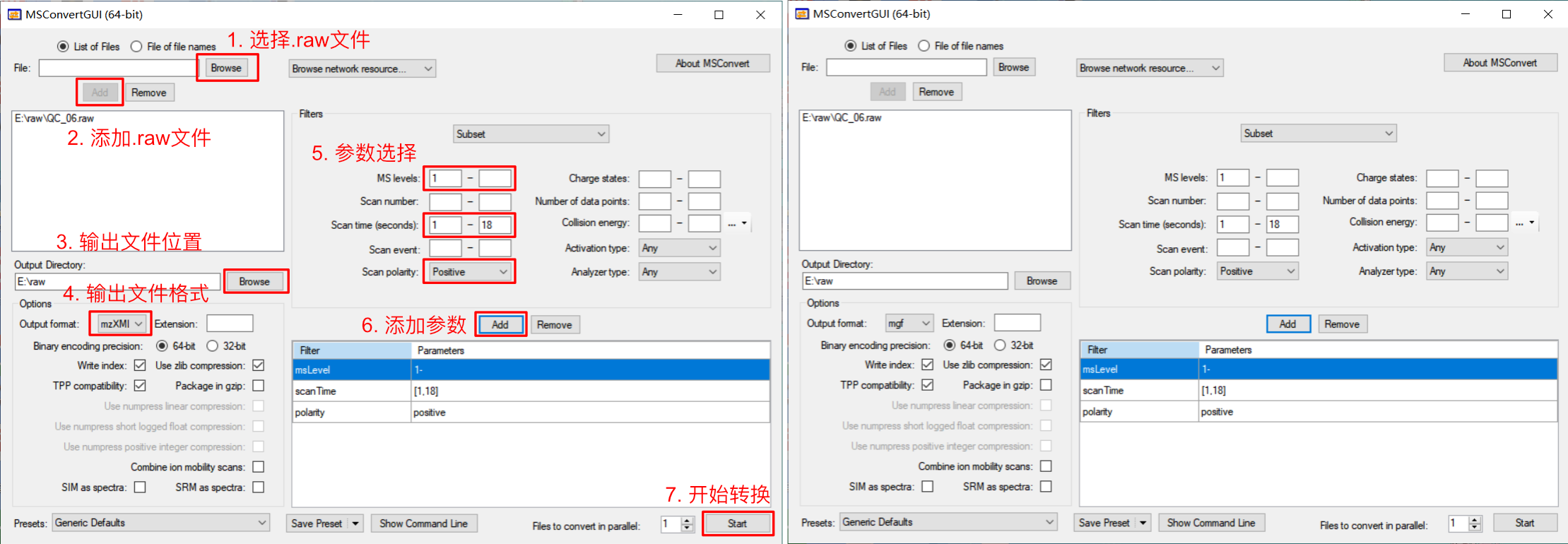 , MSConvert图形化界面软件只可以在windows下运行,MacOS或者linux系统可以根据tidyMass project提供的基于docker的
, MSConvert图形化界面软件只可以在windows下运行,MacOS或者linux系统可以根据tidyMass project提供的基于docker的massconverter的数据转换方式Convert data using massconverter完成原始数据的转换,具体参数及设置参考以上连接。
从原始数据输入
前文提到MetMiner在数据导入时调用了tidyMass project, tidyMass通过xcms对原始数据进行peak picking and grouping
我们这里了解下tidymass对原始数据进行格式转换的过程;
- 首先将LC-MS原始下机数据
.raw格式转换为开源的.mzxml格式的文件,然后读取进来,然后通过massprocesser::process_data()对原始数据进行peak picking and grouping, 获得高可信的特征峰(features),这里定义为variable; - 这些
variable的一级质谱信息m/z(质荷比),rt(保留时间)被记录在variable_info表格中,而根据m/zrt, 这些variable被赋予了例如M92T430_NEG的id,代表该代谢的质荷比为92, 保留时间为430,检出模式为负离子模式; - 此外,根据提峰结果,从不同样本中提取这些variable的峰面积值,这样就形成了一个不同样本中代谢物的积累矩阵,纵向为variable_id, 横向为样本的名称,这个代谢物积累矩阵被称为
expression data; - 同时tidymass还会提供一个初始化的
sample_info,也就是样本信息表,默认的包含了4个主要(必要)的元素,样本名称sample_id,injection order,class,group; 这些在后续会详细说明; - 接下来,通过
.mgf格式的文件将捕获到的ms2 spectra信息读取进来,通过比对代谢物m/z信息和MS2 spectra中的precursor ion信息以及rt信息,通过一定的阈值进行过滤筛选,最终给第一步提峰结果获得的variables填匹配的MS2信息;
下面是一个feature的MS2信息,
BEGIN IONS
TITLE=QC_01.500.500. ## 该feature的ID
RTINSECONDS=60.242274 ## 保留时间(秒)
PEPMASS=146.044738769531 113043348.585899993777 ##(precursor 质荷比 | Intensity)
51.51658919 2715.7590332031 ## 以下为碎裂片段的质荷比和Intensity
57.03285633 7999.3442382813
59.0122181 7026.62109375
61.98706248 182087.59375
70.4917399 4920.7504882813
71.01212618 3894.3942871094
74.02344975 9476.15234375
82.02797575 2661.8305664063
84.04428931 7634.880859375
85.02770882 14845.5263671875
87.00760154 13859.4775390625
98.02348564 4059.72265625
102.0547299 323569.375
103.0582669 9390.1201171875
128.0340961 135462.1875
129.0372687 9628.765625
146.0448944 58732.953125
146.6037865 2932.517578125
147.0289467 7400.3515625
155.2126706 2777.9907226563
160.1064809 2709.7451171875
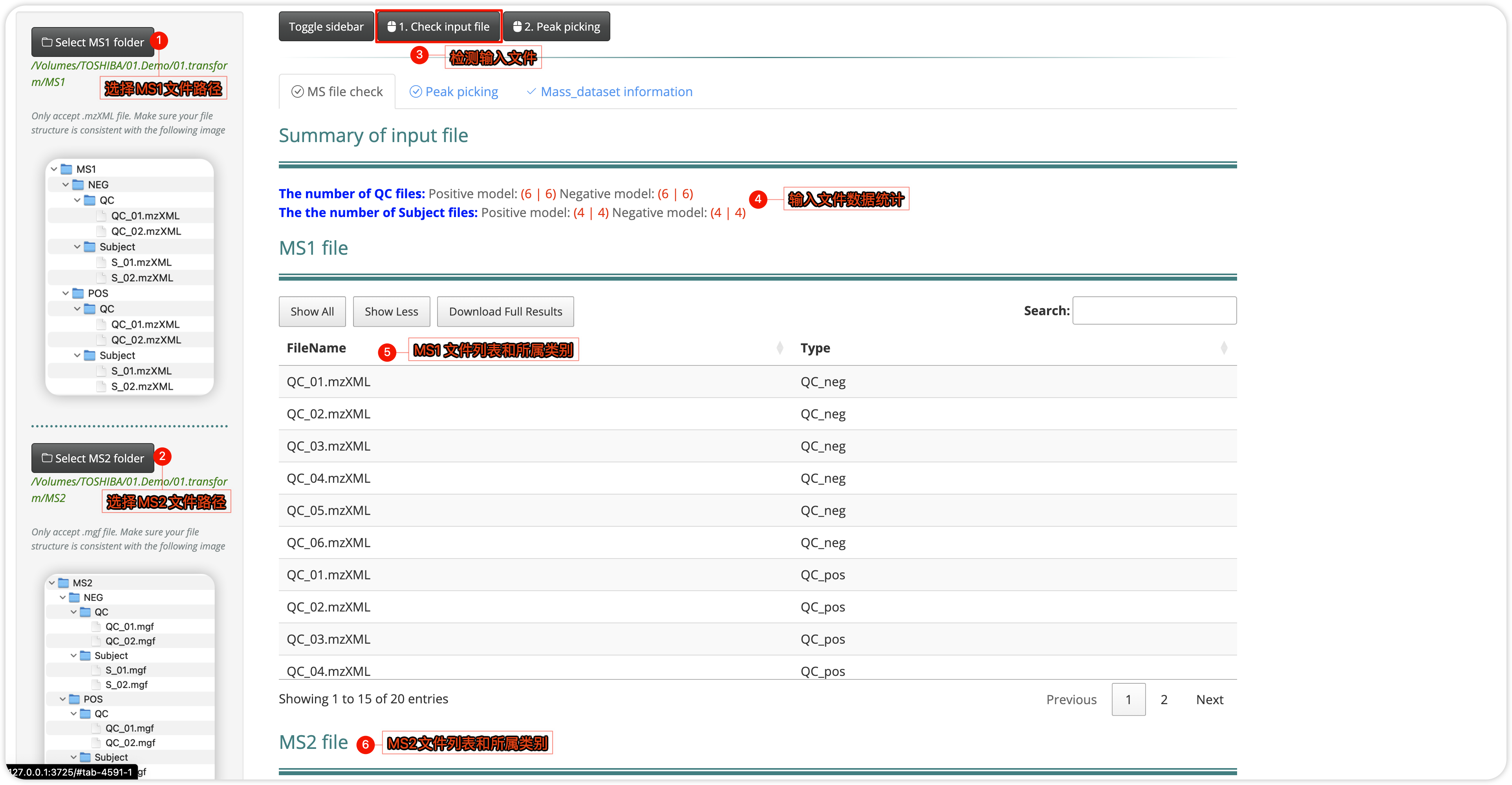
在上集中我们准备好metadata, 组织好数据结构,设置好工作路径后准备将原始数据导入MetMiner,如下图:
- 选则MS1和MS2文件路径;
- 点击
Check input file; - 在MS file check页面检查输入文件统计和列表,看是否有遗漏或者错误文件,对应修改,比如有文件名不符合我们上篇提到的不符合命名规则以及和sample_info表格中不一致的,要做到统一;
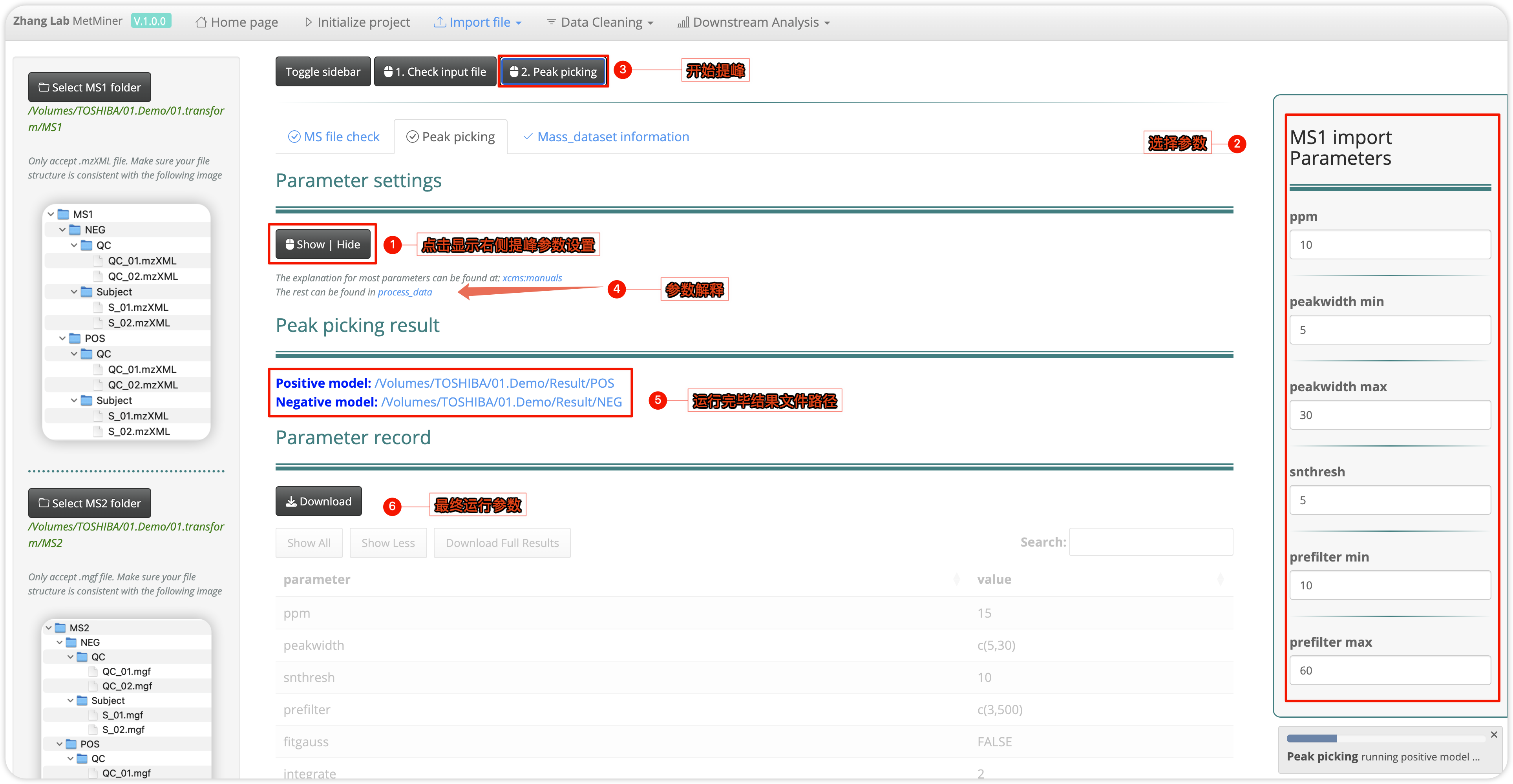
- 点击
Peak picking页面,在parameter setting下面的Show | Hide, 在左边可以弹出提峰参数选择的选项卡,进行提峰参数选择,如果对参数不熟悉,可以点击Show | Hide下面的连接到XCMS和tidyMass网站了解具体参数意义,根据实际情况进行参数选择; - 运行过程中在右下方会有一个progress bar,提醒提峰运行步骤,运行完毕,下面会有提峰结果自动保存路径和参数记录;
--------------------
massdataset version: 1.0.29
--------------------
1.expression_data:[ 4132 x 10 data.frame]
2.sample_info:[ 10 x 4 data.frame]
10 samples:QC_01 QC_02 QC_03 ... S_03 S_04
3.variable_info:[ 4132 x 3 data.frame]
4132 variables:M70T62_POS M70T72_POS M70T921_POS ... M1031T354_POS M1031T353_POS
4.sample_info_note:[ 4 x 2 data.frame]
5.variable_info_note:[ 3 x 2 data.frame]
6.ms2_data:[ 1322 variables x 1207 MS2 spectra]
--------------------
Processing information
3 processings in total
create_mass_dataset ----------
Package Function.used Time
1 massdataset create_mass_dataset() 2024-07-13 10:51:07
process_data ----------
Package Function.used Time
1 massprocesser process_data 2024-07-13 10:49:36
mutate_ms2 ----------
Package Function.used Time
1 massdataset mutate_ms2() 2024-07-13 10:52:39
在完成从原始数据导入后,我们可以在mass_dataset information界面查看到目前质谱数据的状态。这里也直接显示了提峰结果,例如在正离子模式下,
expression_data: 表达量表格,我们获得了4132个代谢物,共有10个sample(包括QC样本);sample_info: 样本信息,包含了10个样本,共有4列信息,包含了
a.sample_id样本id;
b.injection.order上样顺序;
c.class样本分类,分为QC和Subject,说明样本属性,QC代表QC样本,Subject代表测试样本;
d.group样本分组,和前一篇文章提到的一样,这里代表样本的分组,比如case, control,该信息比较重要,是后续噪音去除,PCA,差异分析等的重要依赖信息。variable_info代谢物信息表, 包含4132个代谢物和3列信息:
a.variable_id代谢物id,一般是MxxxTxxx,就是该代谢物的m/z+rt;
b.m/z质荷比,单位为道尔顿;
c.rt保留时间,单位为秒sample_info_note就是介绍sample_info表格中每列的含义;variable_info_note同样,介绍variable_info表格中每列的含义;ms2_data代谢物的ms2信息存储,只有通过mutate_ms2()将peak picking and grouping得到的feature和从.mgf文件中得到的ms2 spectra进行匹配,只有质荷比和保留时间均符合我们设定的tolerance时,才会匹配成功;
从原提峰表格导入
有时候,我们也会从一些经典的提峰软件,比如xcms,CD,mzmine,MS-DIAL等软件获得代谢物积累矩阵表格,那么,我们也可以从提峰表格导入metMiner进行下游分析,这里我们需要从这些提峰软件导出提峰结果中的variable_info 和 expression data两个表格,然后合并成一个表格。如下图:
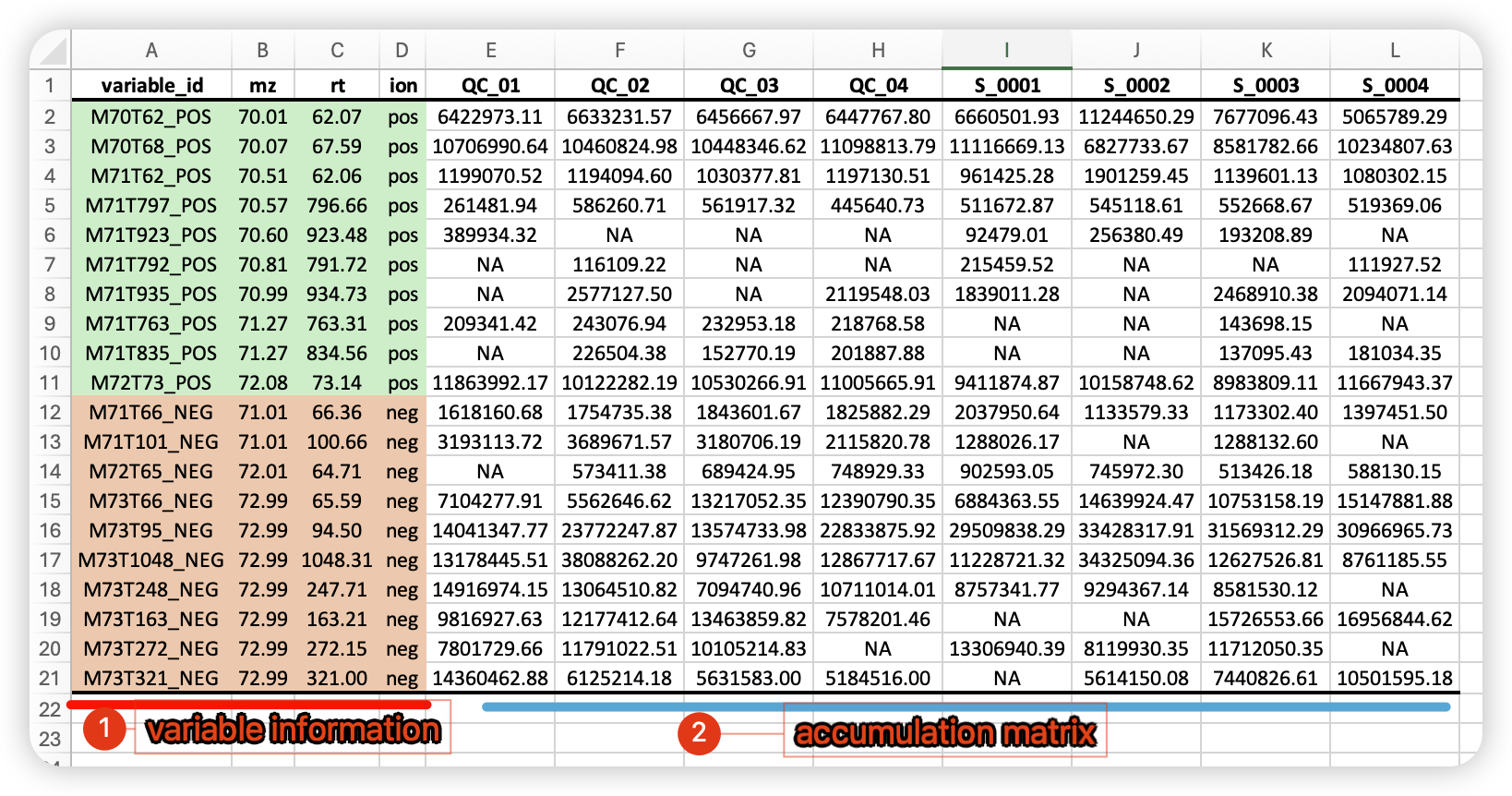
metMiner要求将正负谱检出结果合并到一个表格中,除了variable_id,mz,rt外,还需要加入ion,这里可以是+ -,也可以是pos neg,或者positive negative,软件会自动判断;同时,还需要将原始数据转换过的.mgf文件也准备好,后续同样会进行匹配;
整个数据导入过程如下图:
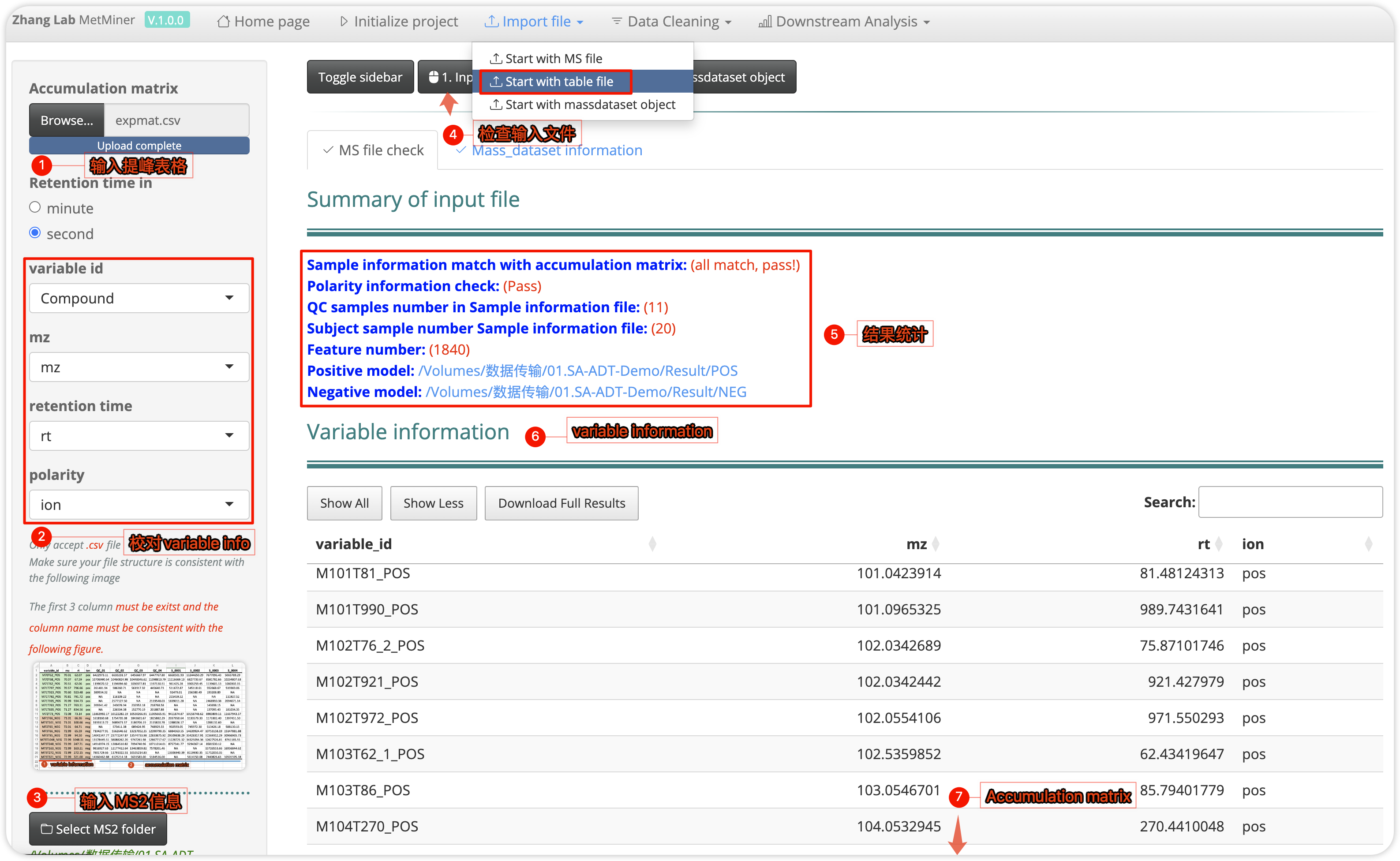
同样,我们设置了variable_info 表格审查,如果你上传的表格中variable_id,mz,rt,ion的列名或者顺序和demo中不一致,需要在这里对应的调整;确保无误,同时上传了MS2文件路径后点击1. Input file summary后,会弹出数据核对信息⑤,variable_info⑥以及Accumulation profile⑦,确保这些信息都无误后,点击2.Generate massdataset object来将质谱数据格式转换为mass_dataset class,此时右下角会出现进度条,这时主要是进行MS2 spectra的匹配; 完成后在Mass_dataset information选项卡中查看mass_dataset打印的信息。
从mass_dataset导入
Tidymass project最大的优点就是质谱数据处理的可重复性、透明性、可追溯性,前面我们通过mass_dataset class的内容就可以体会到透明性、可追溯性,这里从mass_dataset导入也是MetMiner实现可重复性的一种手段。在协作,或者重复已发表数据时,质谱数据已经经过依赖于tidymass的流程处理获得了mass_dataset class类型的质谱数据,我们可以直接加载前人分析的结果重新分析,或者继续下游分析,整个数据导入过程更加简单,只需要获得从R中导入的mass_dataset对象即可,这里我们一般会通过例如save(object_neg, "object_neg.rda")将数据保存为.rda格式,整个操作过程如下图。
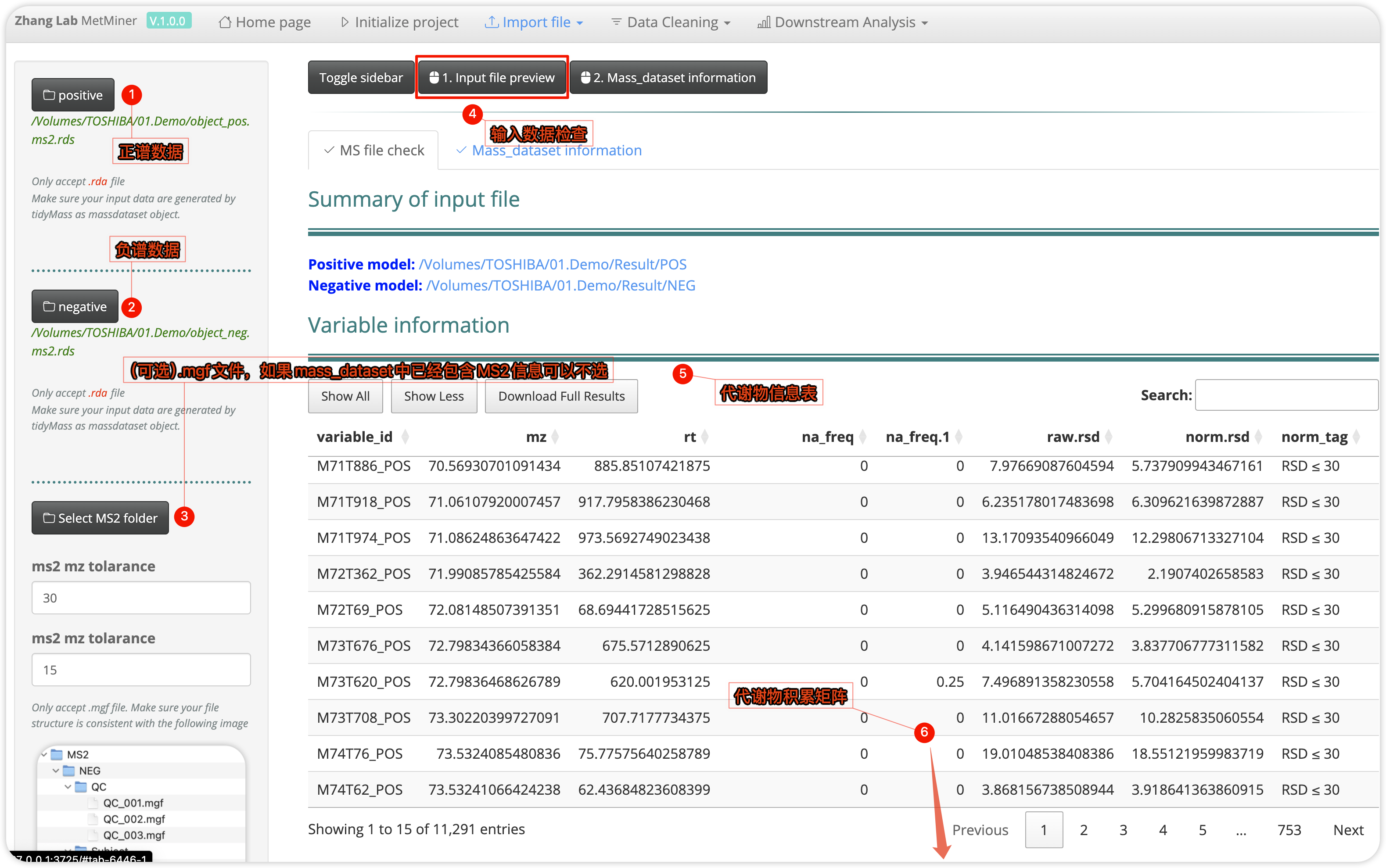
这里需要提醒的是,如果ms2信息在之前的分析中没有添加,才需要再这里上传.mgf文件,如果已经包含,则不需要再次上传;
---The end---
Jul 26, 2024 by Shawn Wang, HENU, Kaifeng, Henan, China.
